clones the repository to a directory of your choice
creates an RStudio project in the cloned directory
updates the .gitignore file
opens the RStudio project in a new RStudio session
Cloning the repository
Initialising {renv}
The {renv} package helps to create reproducible environments for R projects:
records which packages and which versions we are using in the project
isolates the packages library we use in the project from the rest of the computer
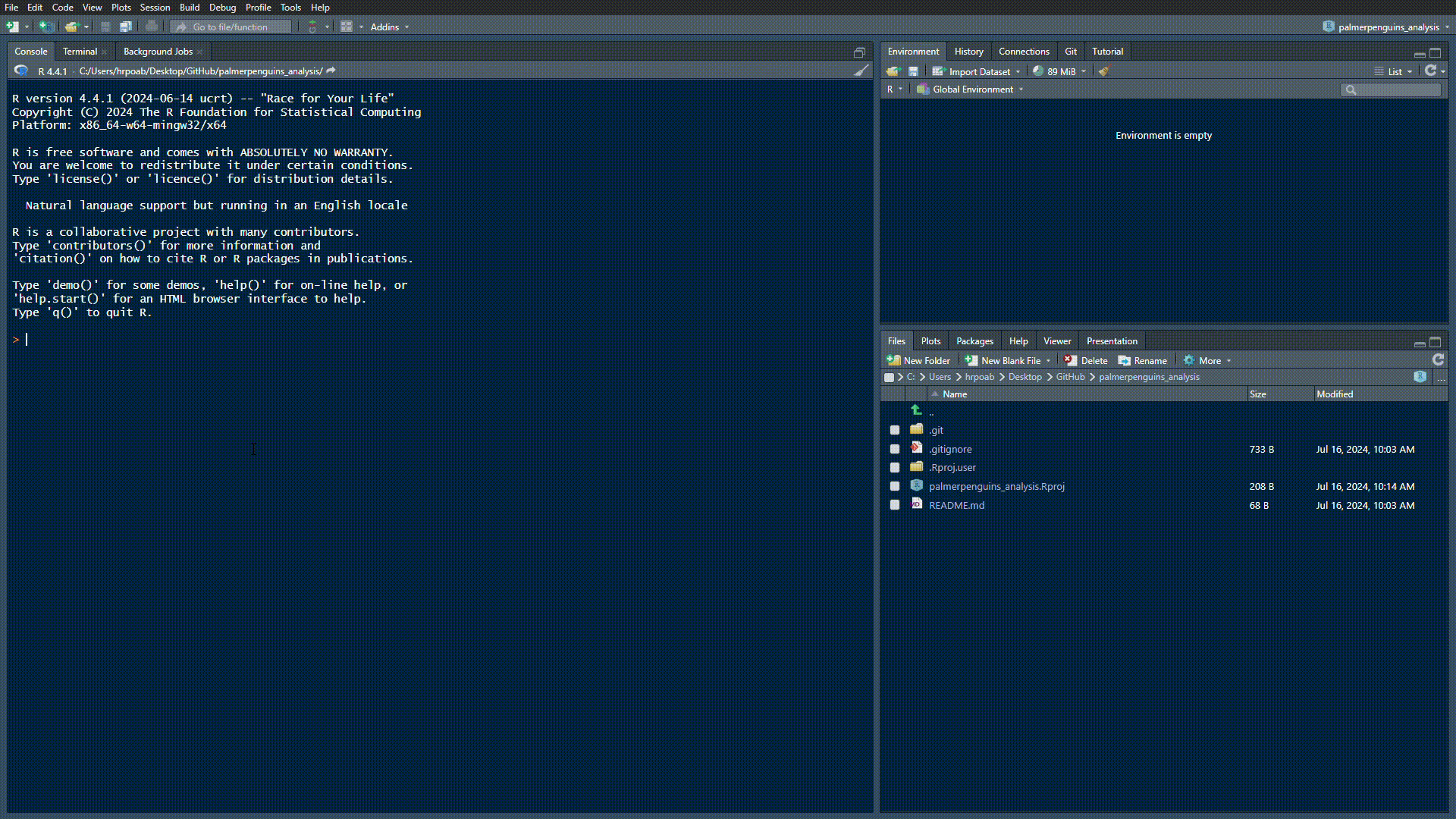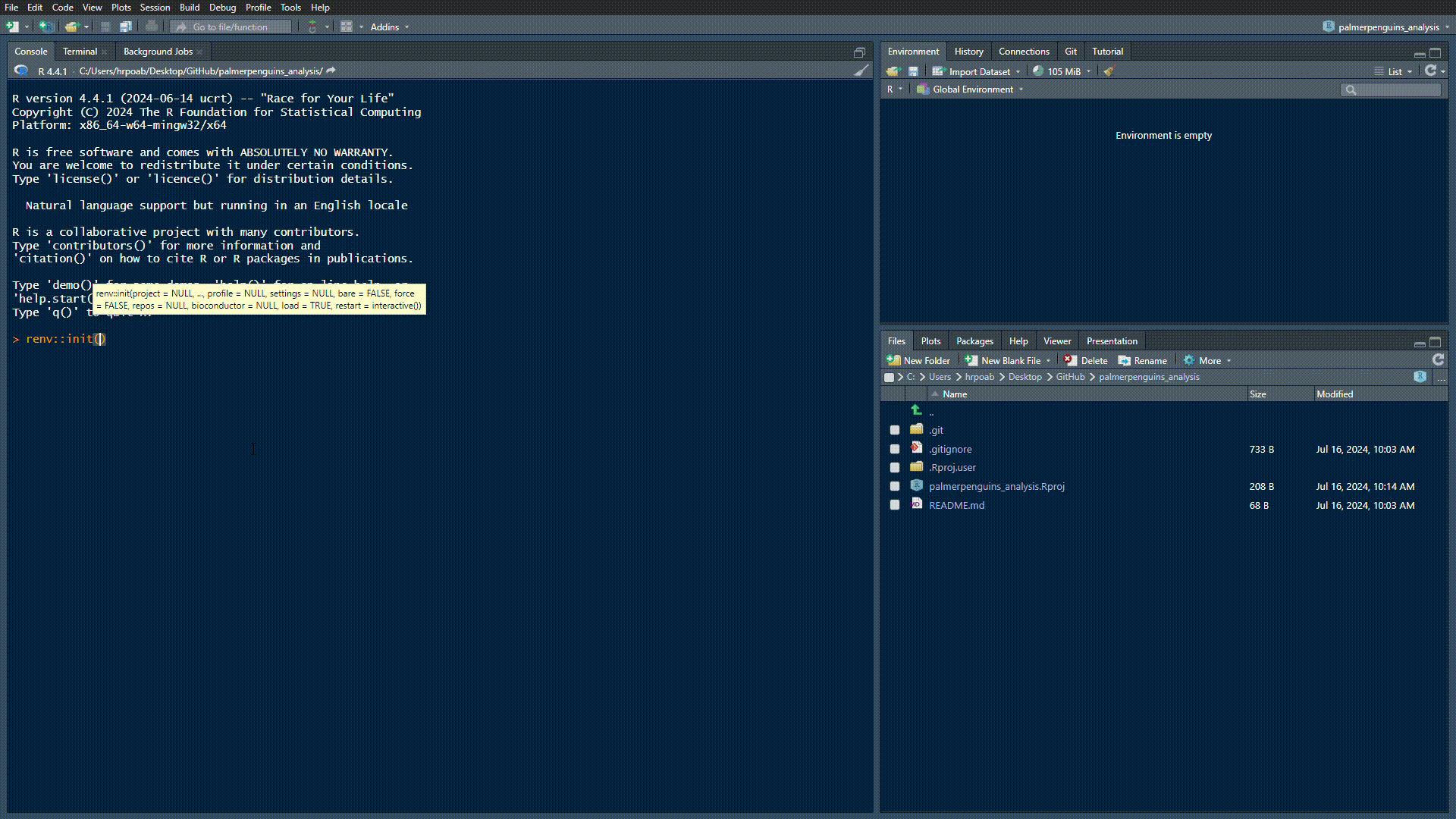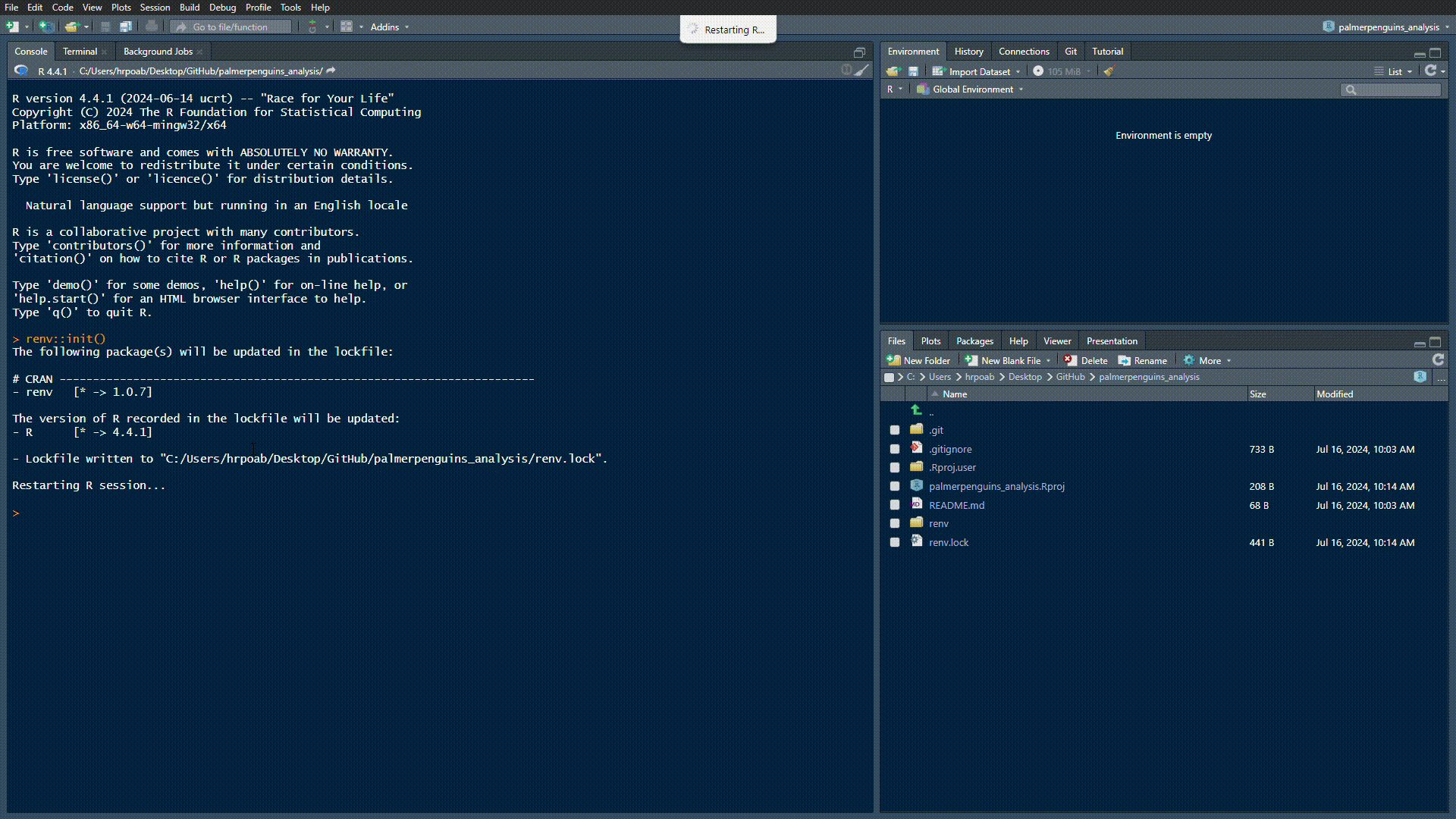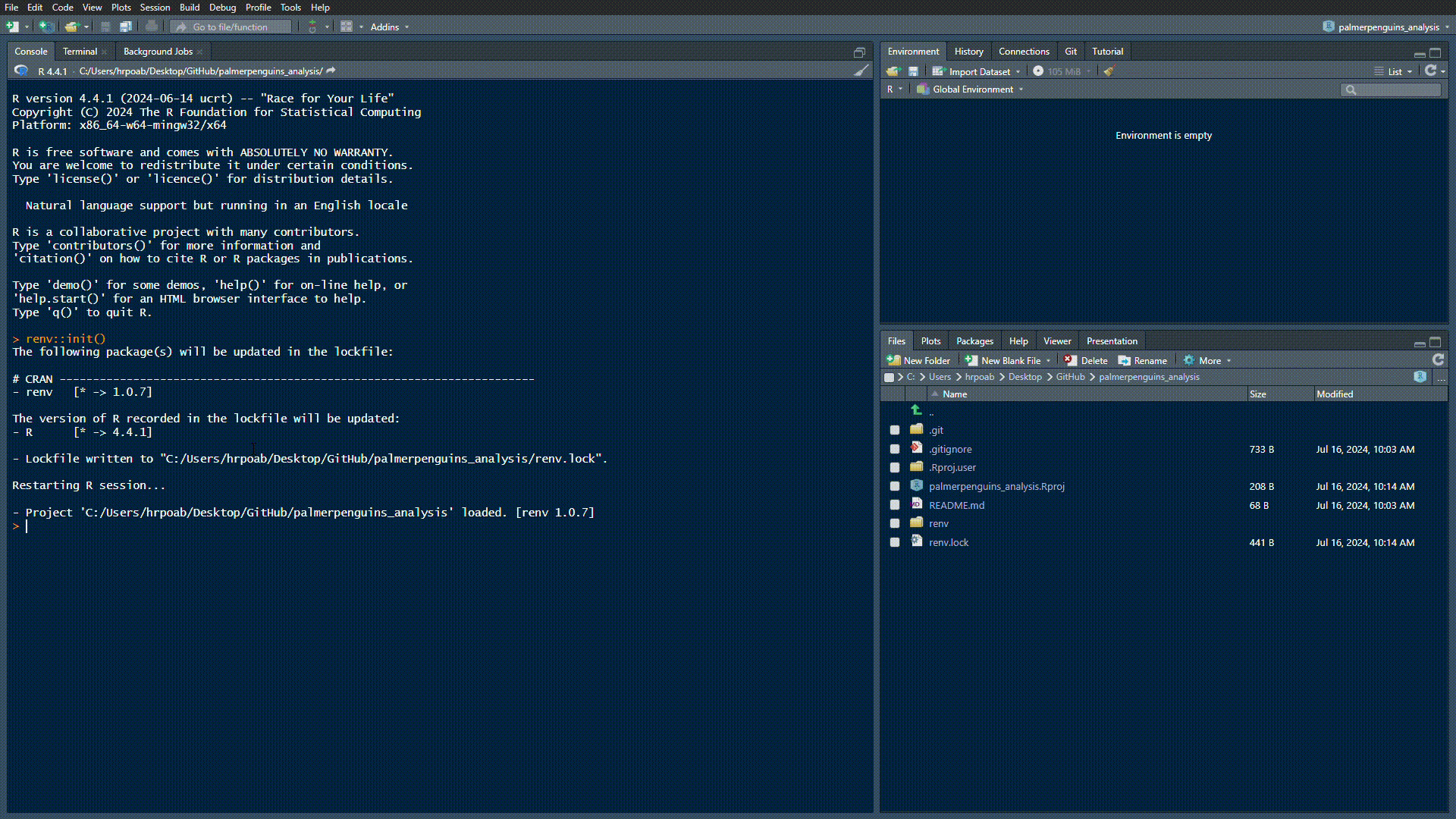
Set-up {renv} in our directory by running renv::init():
creates a renv.lock file that records all dependencies (needs to be tracked on GitHub)
creates a renv/ folder, inside which packages used in the project will be installed (will not be tracked on GitHub)
Initialising {renv}
Using {renv} in four commands
To install a package: renv::install("tidyverse")
Before every commit:
check whether lock file is up-to-date: renv::status()
if not, record the new dependencies: renv::snapshot()
To install dependencies recorded in the lockfile: renv::restore()
Setting up directory structure
Always follows the same organisation – makes it easy for people to understand and find things:
data/: where the data necessary for the project will live (if using local version of data)
analysis/: where to put the analysis scripts
R/: where to put scripts with helper functions
output/: where to save generated tables, figures, etc.
reports/: where to put the files that will be used to generate reports
References
For more information about organising your R project folders, see Marwick et al. (2017) “Packaging Data Analytical Work Reproducibly Using R (and Friends)” and the {rrtools} package.
Setting up directory structure
After creating the folders, here is what our directory looks like:
README should contain a minimal set of information about the project:
a brief description of the project: context, overall aim, some key information about the experiment and the analyses intended/performed
a list of key contributors and their roles, as bullet points
information about the input data: where it is stored, who provided it, what it contains
what analyses were performed, and which files were generated as a result (e.g. cleaned version of the data)
a list of the key folders and files
which sets of commands to run if you want to reproduce the analysis
Populating the README
README.md
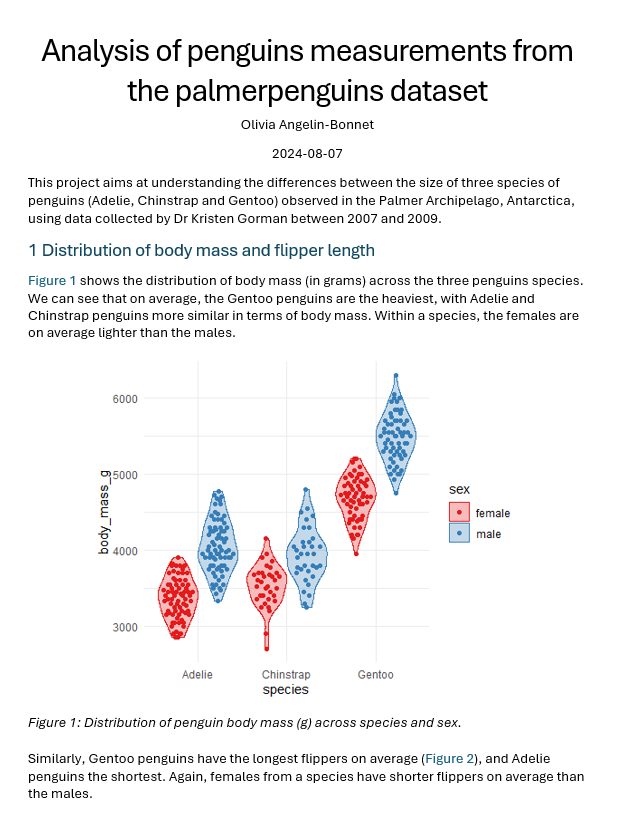
# Analysis of penguins measurementsThis project aims at understanding the differences between the size of three species of penguins (Adelie, Chinstrap and Gentoo) observed in the Palmer Archipelago, Antarctica. The data was collected by Dr KristenGorman between 2007 and 2009.## Key contributors- Jane Doe: project leader- Kristen Gorman: data collection- Olivia Angelin-Bonnet: data analysis## Input dataThe raw penguins measurements, stored in the `penguins_raw.csv` file, were downloaded from the [`palmerpenguins` package](https://allisonhorst.github.io/palmerpenguins/index.html).Data source: [...]## Analysis- Data cleaning: a cleaned version of the dataset was saved as `penguins_cleaned.csv` on[OneDrive](path/to/OneDrive/folder).- *To be filled*## Repository content- `renv.lock`: list of packages used in the analysis (and their version)- `analysis/`: collection of `R` scripts containing the analysis code- `R/`: collection of `R` scripts containing helper functions used for the analysis- `reports/`: collection of `Quarto` documents used to generate the reports## How to reproduce the analysis```{r}# Install the necessary packagesrenv::restore()```
Commit and push
Start coding!
I have written some code…
analysis/first_script.R
library(tidyverse)library(janitor)library(ggbeeswarm)library(here)# Reading and cleaning datapenguins_df <-read_csv(here("data/penguins_raw.csv"), show_col_types =FALSE) |>clean_names() |>mutate(species =word(species, 1),year =year(date_egg),sex =str_to_lower(sex),year =as.integer(year),body_mass_g =as.integer(body_mass_g),across(where(is.character), as.factor) ) |>select( species, island, year, sex, body_mass_g,bill_length_mm = culmen_length_mm,bill_depth_mm = culmen_depth_mm, flipper_length_mm ) |>drop_na()## Violin plot of body mass per species and sexpenguins_df |>ggplot(aes(x = species, colour = sex, fill = sex, y = body_mass_g)) +geom_violin(alpha =0.3, scale ="width") +geom_quasirandom(dodge.width =0.9) +scale_colour_brewer(palette ="Set1") +scale_fill_brewer(palette ="Set1") +theme_minimal()## Violin plot of flipper length per species and sexpenguins_df |>ggplot(aes(x = species, colour = sex, fill = sex, y = flipper_length_mm)) +geom_violin(alpha =0.3, scale ="width") +geom_quasirandom(dodge.width =0.9) +scale_colour_brewer(palette ="Set1") +scale_fill_brewer(palette ="Set1") +theme_minimal()## Scatter plot of bill length vs depth, with species and sexpenguins_df |>ggplot(aes(x = bill_length_mm, y = bill_depth_mm, colour = species, shape = sex)) +geom_point() +scale_colour_brewer(palette ="Set2") +theme_minimal()
I have written some code…
Good start, but often:
many more steps in the analysis → script becomes long and convoluted
harder to get an overview of the analysis, and to find things
don’t want to re-run all the code everytime I make a change
Don’t forget to document your functions! ({roxygen}-style)
#' Read and clean data#' #' Reads in the penguins data, renames and selects relevant columns. The#' following transformations are applied to the data: #' * only keep species common name#' * extract observation year#' * remove rows with missing values#' #' @param file Character, path to the penguins data .csv file.#' @returns A tibble.read_data <-function(file) { readr::read_csv(file, show_col_types =FALSE) |> janitor::clean_names() |> dplyr::mutate(## modifying columns ) |> dplyr::select(## all relevant columns ) |> tidyr::drop_na()}
Step 1: Turn your code into functions
Improved script:
R/helper_functions.R
#' Read and clean data#' #' ...read_data <-function(file) { ... }#' Violin plot of variable per species and sex#' #' ...violin_plot <-function(df, yvar) { ... }#' Scatter plot of bill length vs depth#' #' ...plot_bill_length_depth <-function(df) { ... }
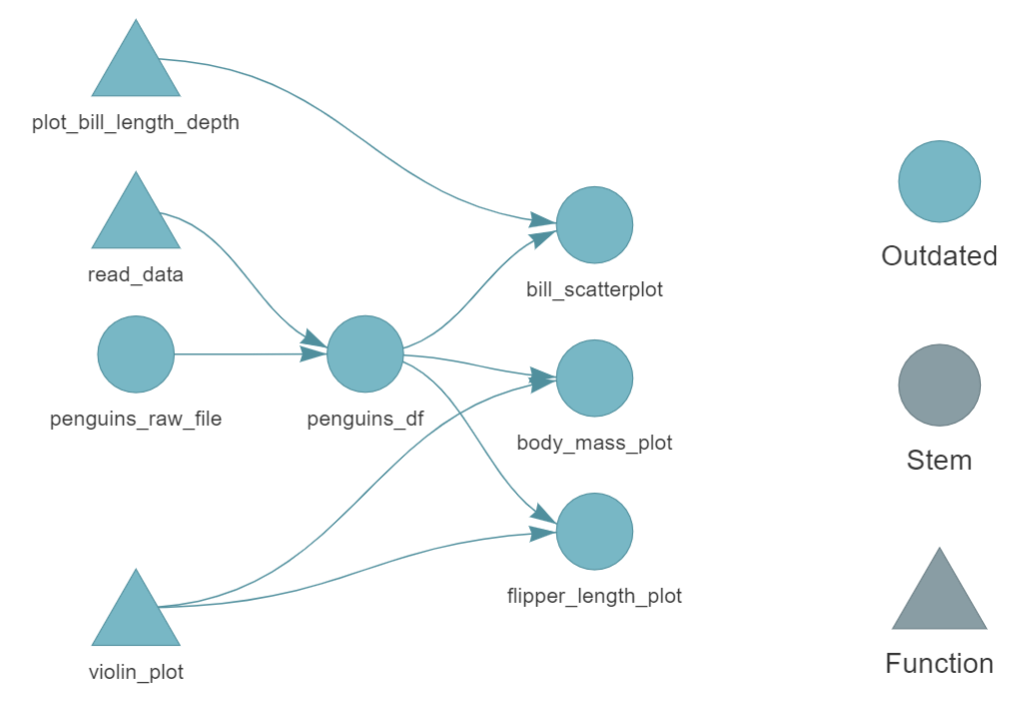
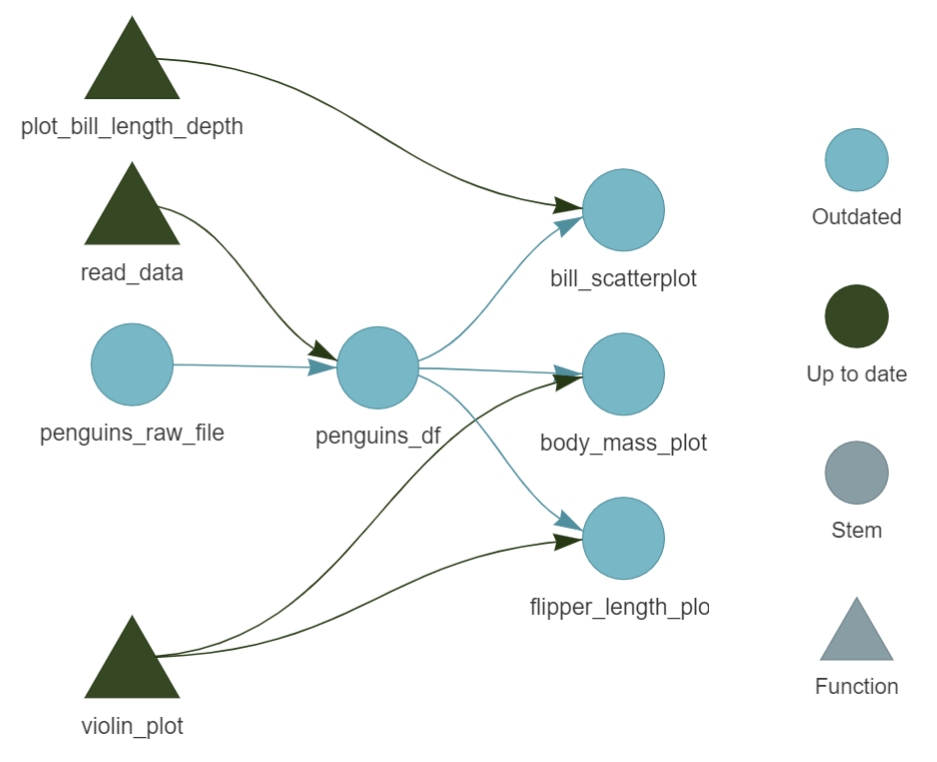
here() starts at C:/Users/hrpoab/Desktop/GitHub/palmerpenguins_analysis
> dispatched target penguins_raw_file
o completed target penguins_raw_file [0 seconds]
> dispatched target penguins_df
o completed target penguins_df [0.85 seconds]
> dispatched target body_mass_plot
o completed target body_mass_plot [0.16 seconds]
> dispatched target bill_scatterplot
o completed target bill_scatterplot [0.02 seconds]
> dispatched target flipper_length_plot
o completed target flipper_length_plot [0.02 seconds]
> ended pipeline [1.31 seconds]
Get the pipeline results
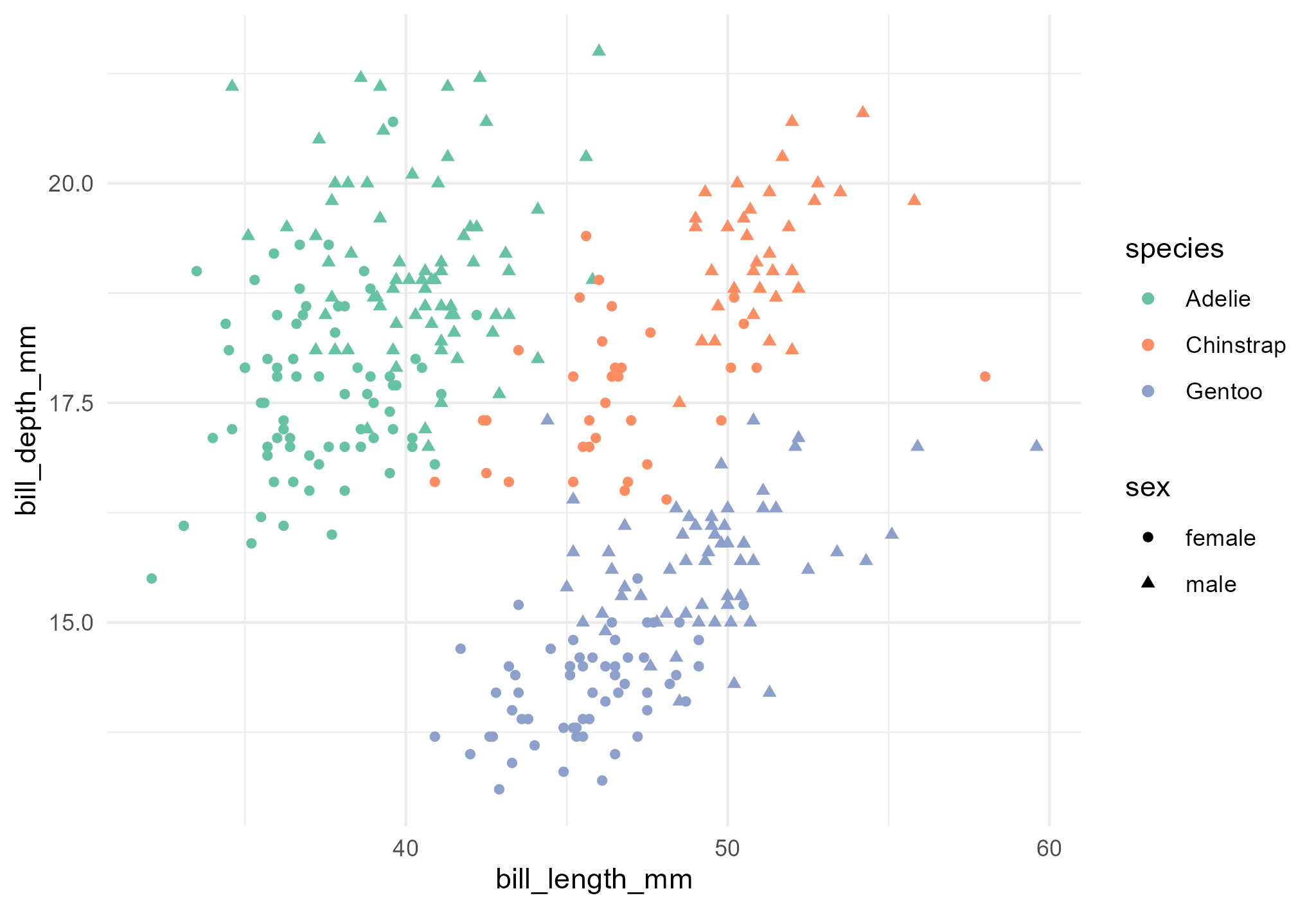
targets::tar_read(bill_scatterplot)
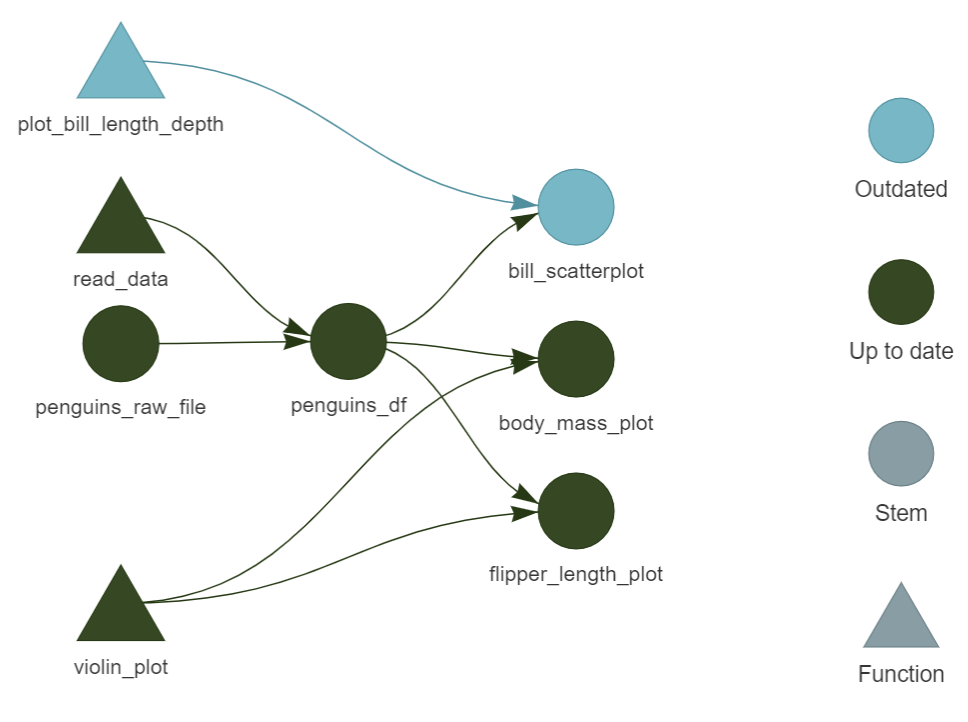
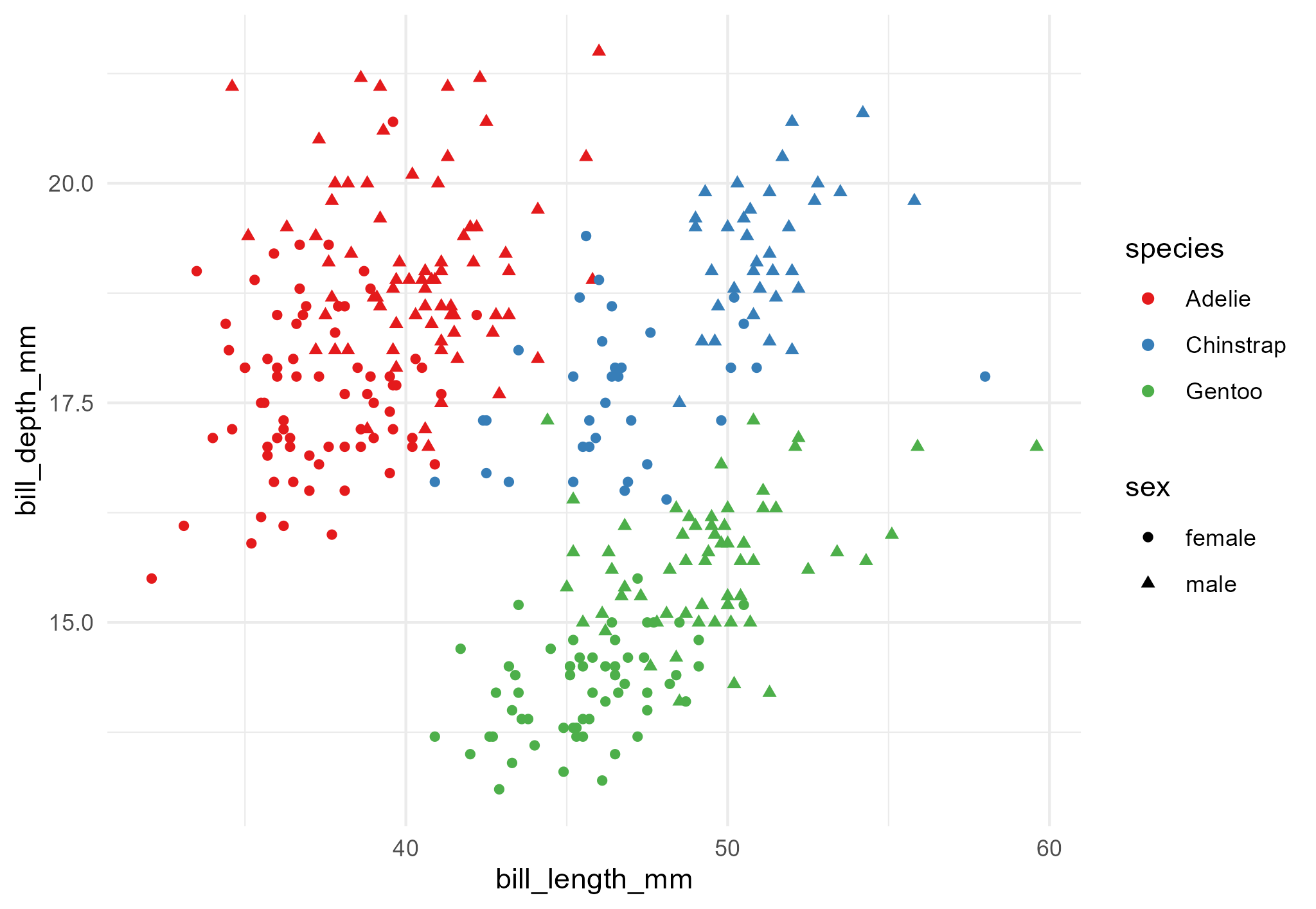
Change in a step
Hi Olivia,
Great work! Just a minor comment, could you change the colours in the bill length/depth scatter-plot? It’s hard to see the difference between the species.
Great work! Just a minor comment, could you change the colours in the bill length/depth scatter-plot? It’s hard to see the difference between the species.
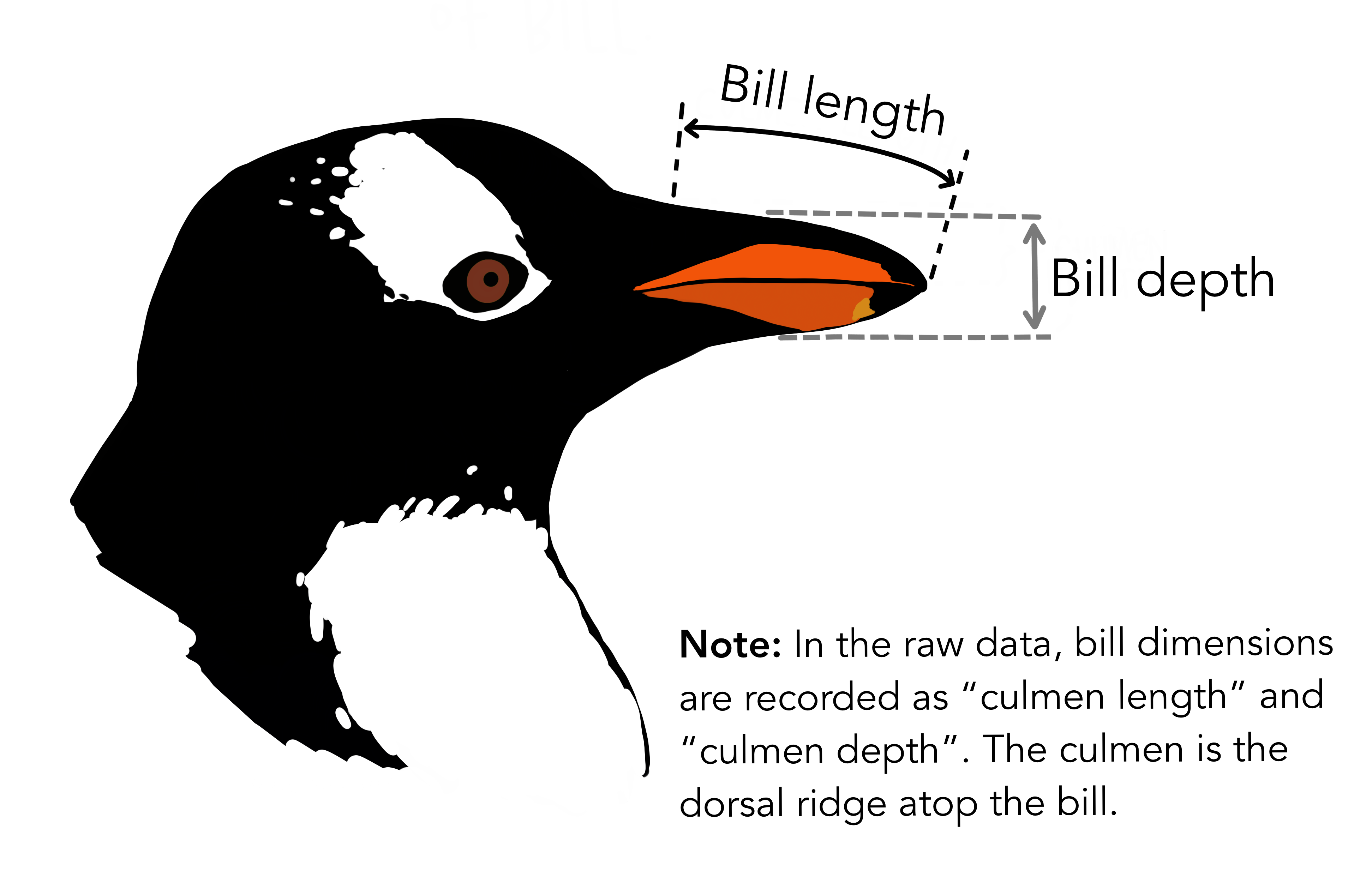
---title: "Analysis of penguins measurements from the palmerpenguins dataset"author: "Olivia Angelin-Bonnet"date: todayformat: docx: number-sections: true---```{r setup}#| include: falselibrary(knitr)opts_chunk$set(echo = FALSE)```This project aims at understanding the differences between the size of three species of penguins (Adelie, Chinstrap and Gentoo) observed in the Palmer Archipelago, Antarctica, using data collected by Dr Kristen Gorman between 2007 and 2009.## Distribution of body mass and flipper length@fig-body-mass shows the distribution of body mass (in grams) across the three penguins species. We can see that on average, the Gentoo penguins are the heaviest, with Adelie and Chinstrap penguins more similar in terms of body mass. Within a species, the females are on average lighter than the males.```{r fig-body-mass}#| fig-cap: "Distribution of penguin body mass (g) across species and sex."# code for plot```Similarly, Gentoo penguins have the longest flippers on average (@fig-flipper-length), and Adelie penguins the shortest. Again, females from a species have shorter flippers on average than the males.```{r fig-flipper-length}#| fig-cap: "Distribution of penguin flipper length (mm) across species and sex."# code for plot```## Association between bill length and depthIn this dataset, bill measurements refer to measurements of the culmen, which is the upper ridge of the bill. There is a clear relationship between bill length and depth, but it is masked in the dataset by differences between species (@fig-bill-scatterplot), with Gentoo penguins exhibiting longer but shallower bills, and Adelie penguins shorter and deeper bills.```{r fig-bill-scatterplot}#| fig-cap: "Scatterplot of penguin bill length and depth."# code for plot```
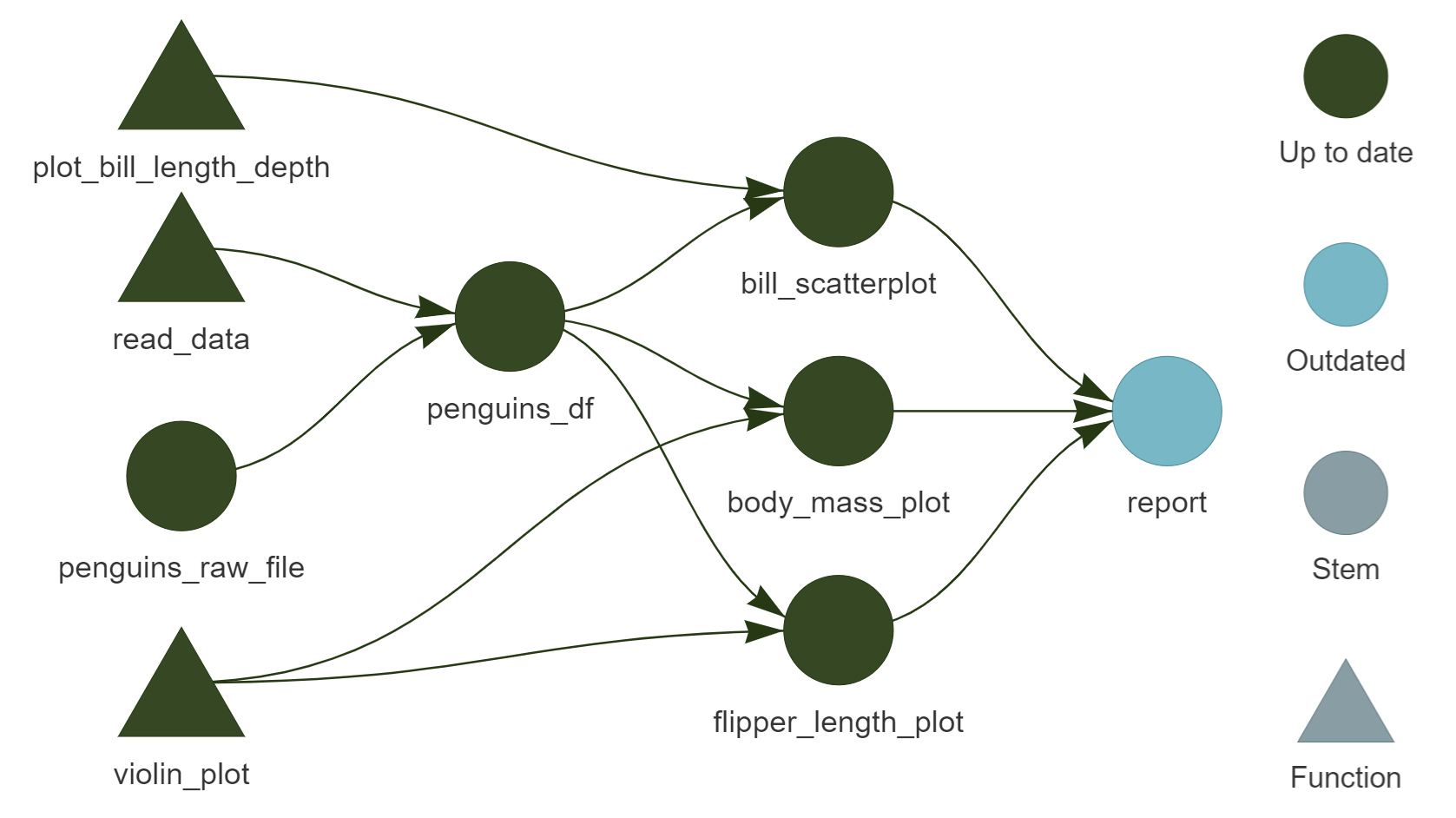
Writing a report – Quarto + {targets}
Two advantages of using a Quarto document alongside {targets}:
can read in results from targets pipeline inside the report: no computation done during report generation
can add the rendering of the report as a step in the pipeline: ensures that the report is always up-to-date
Writing a report – Quarto + {targets}
reports/palmerpenguins_report.qmd
---title: "Analysis of penguins measurements from the palmerpenguins dataset"author: "Olivia Angelin-Bonnet"date: todayformat: docx---```{r setup}#| include: falselibrary(knitr)opts_chunk$set(echo = FALSE)```This project aims at understanding the differences...## Distribution of body mass and flipper length@fig-body-mass shows...```{r fig-body-mass}#| fig-cap: "Distribution of penguin body mass (g) across species and sex."# code for plot```
Two steps to use the {targets} pipeline results in a Quarto document:
Writing a report – Quarto + {targets}
reports/palmerpenguins_report.qmd
---title: "Analysis of penguins measurements from the palmerpenguins dataset"author: "Olivia Angelin-Bonnet"date: todayformat: docx---```{r setup}#| include: falselibrary(knitr)library(here)opts_chunk$set(echo = FALSE)opts_knit$set(root.dir = here())```This project aims at understanding the differences...## Distribution of body mass and flipper length@fig-body-mass shows...```{r fig-body-mass}#| fig-cap: "Distribution of penguin body mass (g) across species and sex."# code for plot```
Two steps to use the {targets} pipeline results in a Quarto document:
Make sure the report ‘sees’ the project root directory
Writing a report – Quarto + {targets}
reports/palmerpenguins_report.qmd
---title: "Analysis of penguins measurements from the palmerpenguins dataset"author: "Olivia Angelin-Bonnet"date: todayformat: docx---```{r setup}#| include: falselibrary(knitr)library(here)library(targets)opts_chunk$set(echo = FALSE)opts_knit$set(root.dir = here())```This project aims at understanding the differences...## Distribution of body mass and flipper length@fig-body-mass shows...```{r fig-body-mass}#| fig-cap: "Distribution of penguin body mass (g) across species and sex."tar_read(body_mass_plot)```
Two steps to use the {targets} pipeline results in a Quarto document:
Make sure the report ‘sees’ the project root directory
Read targets objects with targets::tar_read()
Writing a report – Quarto + {targets}
Adding the Quarto report as a step in the pipeline (need {tarchetypes} and {quarto}):
Presentation for ECSSN and NZSA Joint Webinar, 20 August 2024
Publication data: Angelin-Bonnet O. September 2024. Setting up a reproducible data analysis project in R featuring GitHub, {renv}, {targets} and more. A Plant & Food Research PowerPoint presentation. SPTS No. 26044.
Presentation prepared by: Olivia Angelin-Bonnet Scientist, Data Science September 2024
Presentation approved by: Mark Wohlers Science Group Leader, Data Science September 2024
For more information contact: Olivia Angelin-Bonnet DDI: +64 6 355 6156 Email: Olivia.Angelin-Bonnet@plantandfood.co.nz
This report has been prepared by The New Zealand Institute for Plant and Food Research Limited (Plant & Food Research). Head Office: 120 Mt Albert Road, Sandringham, Auckland 1025, New Zealand, Tel: +64 9 925 7000, Fax: +64 9 925 7001. www.plantandfood.co.nz
DISCLAIMER
The New Zealand Institute for Plant and Food Research Limited does not give any prediction, warranty or assurance in relation to the accuracy of or fitness for any particular use or application of, any information or scientific or other result contained in this presentation. Neither The New Zealand Institute for Plant and Food Research Limited nor any of its employees, students, contractors, subcontractors or agents shall be liable for any cost (including legal costs), claim, liability, loss, damage, injury or the like, which may be suffered or incurred as a direct or indirect result of the reliance by any person on any information contained in this presentation.
Setting up a reproducible data analysis project in R featuring GitHub, {renv} , {targets} and more Olivia Angelin-Bonnet The New Zealand Institute for Plant and Food Research 20 August 2024